ここ数日、SAM/BAMファイルのcigar列の読み方で悩んでいて、色々と調べたりぐぐったり人に聞いたり、とやっておりました。
#というか、seqanswerとかでもその手のやりとりがあったりして、結構悩んでいる人は居るのだなぁ、とか思ったり。
正直ちゃんと完全に理解できたかどうか自信が無い部分もあるのですが、一応今の段階で調べたことをメモっておきたいと思います。
とにもかくにもマニュアルを見てみる。
https://samtools.github.io/hts-specs/SAMv1.pdf
5ページ目を見てみます。
M 0 alignment match (can be a sequence match or mismatch)
I 1 insertion to the reference
D 2 deletion from the reference
N 3 skipped region from the reference
S 4 soft clipping (clipped sequences present in SEQ)
H 5 hard clipping (clipped sequences NOT present in SEQ)
P 6 padding (silent deletion from padded reference)
= 7 sequence match
X 8 sequence mismatch
このそれぞれが分かればいい、と。
ちなみにマニュアルの1ページ目にはexampleが載っています。これらを見比べていくことにしましょう。
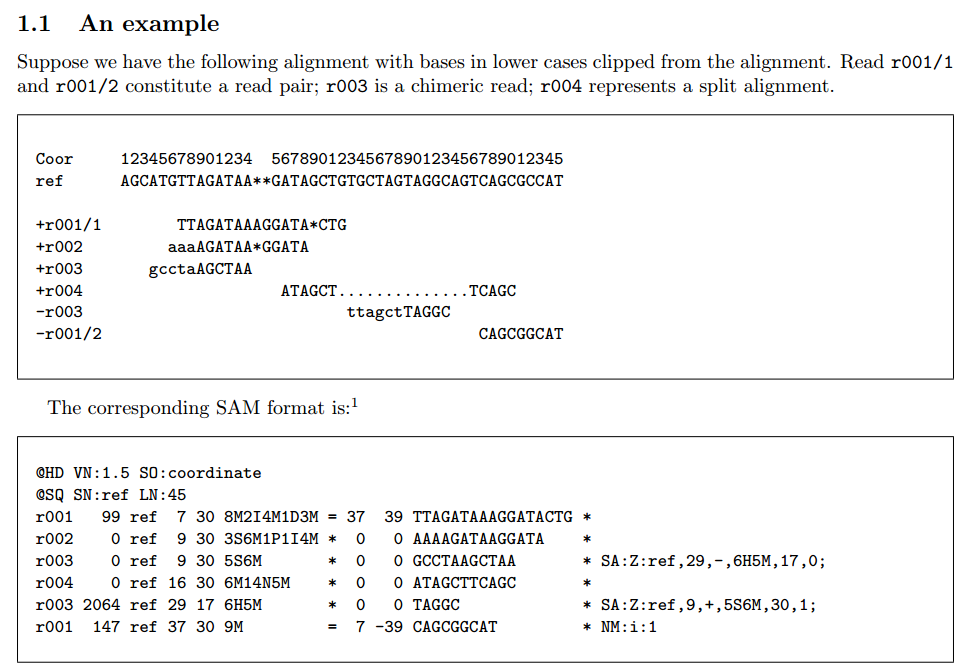
(https://samtools.github.io/hts-specs/SAMv1.pdfよりキャプチャ)
M 0 alignment match (can be a sequence match or mismatch)
“alignment matchとsequence match or mismatch”の違いでいきなり混乱しました。結論としては、alignment matchというのは参照配列にリードを張り付けたよ、ということ意味するだけで、その中身の個々の塩基がmatchしているかどうかは書いていないよ、ということらしく。
現に、exampleの+r003ですが、
参照配列:ATGTTAGATAA
+r003:gcctaAGCTAA
となっていて、これが5S6Mとなっています。
5Sの部分はあと回しにして6Mですが、参照配列はAGATAA、r003はAGCTAAで中身が微妙に異なっています。
おそらくこの6Mは、2=1X3=とも書ける…のかな?
とある人に聞いた限りでは、そもそもxと=は(その人の経験では)あまり見ない、とも言っていたのですが。
I 1 insertion to the reference
D 2 deletion from the reference
IとDはでそれぞれ挿入と欠失、あわせてインデルですね。これは見たまんまなのでまぁ、わかります。
N 3 skipped region from the reference
これが地味に悩みました。skipって、deletionと何が違うの? って。
でもって、ちゃんとマニュアルに補足が。
For mRNA-to-genome alignment, an N operation represents an intron. For other types of alignments,
the interpretation of N is not defined.
要は、mRNAのデータならば、intronである、と。他の場合はマニュアルでは定義されていないのでデータが出てこない限りは考えなくていい、と。
なるほどなるほど。
さて、次。
S 4 soft clipping (clipped sequences present in SEQ)
H 5 hard clipping (clipped sequences NOT present in SEQ)
ここも躓きました。
知ってる人には当たり前のネタなのでしょうが、そもそもソフトクリップとかハードクリップとかって何ぞ? と。
結論としては、soft clip、hard clip共に、貼付けられたリードのうち、レファレンスには張り付かない部分と判断された両端の部分を指す、らしく。
そのうち、SAMファイル上に情報が明示されているものがsoft clip、そもそも何が張り付いていないのか良くわからないのがhard clip、と。
日本語で解説を書いてらっしゃる方もいらっしゃいました。
http://qiita.com/usuyama/items/2338cb7f75aa9407a1c2
Soft clipはsequenceの項に含まれているがalignしなかったもの、hard clipはalignせずsequenceの項にも含まれていない部分を指す。soft clipped readsはIGVで見るとミスマッチが連なって見える。
上記サイトのIGVの画像のリンク先は見つからなかったので、後日自分でも確認できたらブログにUPでもしたいと思います。
ちなみにマニュアルのPDFには、
H can only be present as the first and/or last operation.
S may only have H operations between them and the ends of the CIGAR string
とありました。ハードクリップーソフトクリップーリード張り付きーソフトクリップーハードクリップ、という並びもありうる、ということかな?
さて、ちなみにここですが、ちょっと混乱したのが、chimeric readという言葉でした。先ほど転載したリード例の図の中に、
r003 is a chimeric read
とあり、これが何なのだろう、と。
一応、マニュアルの中に
Chimeric alignment An alignment of a read that cannot be represented as a linear alignment. A chimeric
alignment is represented as a set of linear alignments that do not have large overlaps. Typically, one
of the linear alignments in a chimeric alignment is considered the “representative” alignment, and the
others are called “supplementary” and are distinguished by the supplementary alignment flag.
などとあり、ああ、何か違った配列がひっついたものなんだね、だからソフトクリップとかになるんだね、というのは分かるのですが、ちょっとすっきりしなかったというか。いわゆるキメラ遺伝子(chimeric gene)とは違うのかな? とか。
biostarsにも似たようなやりとりがあったり、と。
https://www.biostars.org/p/116201/
で、上記記事を見てみたり、あと人に聞いたりして自分なりに把握したのは、chimeric readとは、いわゆるfusion geneみたいな、実際の配列上に出てくる融合遺伝子やその他の変異だけでなく、pcrのエラーとかで出てくるものも含む、ということ。
だから、chimeric readとは実際の遺伝子以外の場合もありうる、ということかな、と。なるほどなるほど。
さて、最後にpaddingです。
P 6 padding (silent deletion from padded reference)
これも混乱しました。というか混乱してばっかですね。”silent deletion from padded reference”とあり、またマニュアルの例の図(002のリード)張り付いたリードから何か欠失が起こっている、ということを意味するのかな、というのは何となくわかったのですが、あまり知らない話だったので実感がわかなかった、というか。
で、いろいろググって、以下のような記述にたどり着きました。
http://davetang.org/wiki/tiki-index.php?page=SAM
Padded alignment is always produced by de novo assemblers and is important for an alignment viewer to display the alignment properly. To store padded alignment, we introduce operation ‘P’ which can be considered as a silent deletion from padded reference sequence.
なるほど、de novoメインの話なんですね。
リードをどう読むか、ということを含めて一応納得。
=とXについてはさっきやりましたね。
という訳でこれで一通り理解した、ということになったかな、と。
自分なりにいちおうスッキリ、という感じなのでした。
さて、まとめます。
| M | 参照配列に張り付いたリード。ただし、個々の塩基が参照配列と同じかどうかは感知しない。 |
| I | 挿入。 |
| D | 欠失。 |
| N | 間が飛んでいる。mRNAではイントロンがメイン。 |
| S | ソフトクリップ。張り付いていない端っこのリード。何が張り付いていないのかは明記されている。 |
| H | ハードクリップ。張り付いていない端っこのリード。何が張り付いていないのかは不明。 |
| P | padding。張り付いたリードの側で何かの欠失が起こっている。de novoがメイン? |
| = | 参照配列に張り付いたリードのうち、参照配列と同じ塩基。 |
| X | 参照配列に張り付いたリードのうち、参照配列とは違う塩基。 |
……ふぅ。とりあえずスッキリしました。また間違い等があった場合は修正していきたいと思います。
※その他、参考にさせて頂いたページ
The Sequence Alignment/Map format and SAMtools
http://crusade1096.web.fc2.com/sam.html
http://seqanswers.com/forums/showthread.php?t=4882